A practical titration guide if you want to learn the difference between automated and manual titrations and the advantages of both methods, need help figuring out which method is right for your particular application, or are looking for tips on improving volume accuracy. The practical guide provides insights into the basics, methods and application areas of titration.
The Titration handbook combines application information with the practical laboratory experience of our titration experts. This guide requires chemical knowledge, e.g. the reading of reaction equations, knowledge of important technical terms, basic knowledge of working in the chemical laboratory, as well as the handling of devices such as scales, burettes, pipettes, electrodes and the safety regulations in the laboratory.
Practical titration guide
 Titration is one of the oldest methods for content determination in chemistry. On the one hand, a titration can be performed very easily and quickly. On the other hand, titration provides a very accurate measurement result after only a few minutes - under optimal conditions. A relative standard deviation of less than one percent is normal. Not without reason do numerous standards require titration as a method. This explanation of titration builds on the general principles of titration and is aimed at the user of potentiometric titration. Therefore, the basics of potentiometry with Nern's equation are dealt with.
Titration is one of the oldest methods for content determination in chemistry. On the one hand, a titration can be performed very easily and quickly. On the other hand, titration provides a very accurate measurement result after only a few minutes - under optimal conditions. A relative standard deviation of less than one percent is normal. Not without reason do numerous standards require titration as a method. This explanation of titration builds on the general principles of titration and is aimed at the user of potentiometric titration. Therefore, the basics of potentiometry with Nern's equation are dealt with.
In the first step, this titration guide deals with the volume and its correctness. Thereafter, the focus is on the sample and its handling. Subsequently, the used reagents, electrodes and the titration parameters are dealt with in detail.
Titration reactions
The definition of titration is valid unchanged in its core: We need a stoichiometric reaction, a precisely dosable, stable reagent and a detection of the end of the reaction end or a curve showing the course of the reaction.

- The chemical reaction on which the titration is based must proceed rapidly, quantitatively and unambiguously in the manner indicated by the reaction equation.
- It must be possible to prepare a reagent solution of defined concentration or to determine the concentration of the solution in a suitable way.
- The endpoint of the titration must be clearly recognizable. It should coincide with the equivalence point at which the reagent amount equivalent to the substance amount of the searched substance was added or at least come very close to it.
A simple and very reliable method is acid-base titration, which has been used for a very long time to determine the concentration of aqueous solutions. In acid-base or neutralization titration, acids are titrated with a base (or vice versa). The detection of the equivalence point can take place by colour indicators or potentiometrically with a glass electrode. The reaction is the same for all acid/base titrations, water results from a proton and a hydroxide ion. Acid-base titrations are mainly in use: for monitoring environmental processes, such as the analysis of water and soil quality, but also in the production of food and pharmaceuticals.
Comparison of manual and automatic titration
Even if manual titrations are still carried out today, the many advantages speak in favor of using an automatic titrator.

Fig. 1: Modern titrator TitroLine® with sample changer in use: TitroLine TL 7800-M1 (Titrations-Fibel Page 46).
Fig. 2 compares the two forms of application.Manual titration is often faster. The duration of the titration consists of the reaction duration and the setting of the sensor potential.
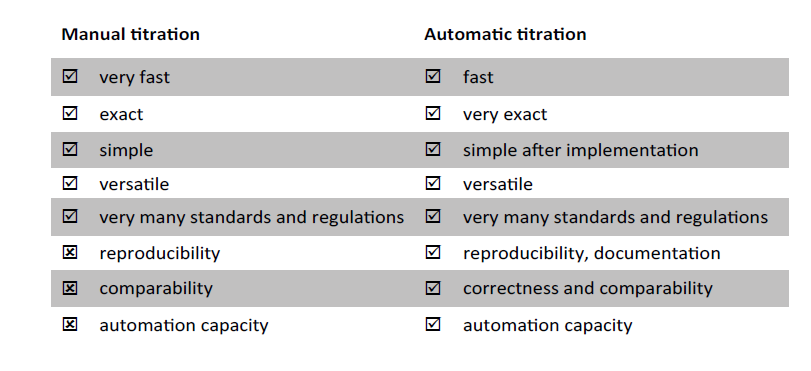
Fig. 2: Comparison of manual and automatic titration.
With manual titration, the sensor and thus its influence on the total titration time are eliminated. In slow reactions, too fast manual titration can lead to over- or under- findings. Thus, some redox reactions are so slow that they run at higher temperatures or that catalysts must be added. There is a risk here that the titration in manual water determination is too fast and the chemical conversion cannot follow.
The automatic potentiometric titration works in a drift-controlled manner, that is, the progress of the reaction can be monitored by means of a sensor. With manual titration with indicator, one titrates until the colour changes. With the content calculation, exactly one point of the entire titration is thus used for the evaluation.
There thus does not exist any possibility to get information about:
- Reaction process
- Signal/noise ratio
- Behavior directly before and
- after the endpoint
- Characteristics for an uncertainty consideration
- Are there several endpoints?
With the potentiometric titration, the entire titration curve is available and thus also evaluation criteria for the above points. In addition, several measurement points in the region of the equivalence point are used for an equivalence point calculation.
With the endpoint titration, one also titrates to one point. The direct potentiometric implementation of a manual titration is thus an endpoint titration to the point where the indicator changes colour. In contrast to manual titration, a complete titration curve is also available here for evaluation.
In Fig. 3, the equivalence point is at pH 7.42. The colour change starts at pH 6.80, an endpoint titration would be ended at pH 7.00. However, as the titration curve is very steep, differences in consumption, which is in effect the measurement unit of the titration, are very low. For flat titration curves, in contrast, significant differences can occur. The three different detections (optical manual, EP titration and EQ titration) must therefore be regarded as (slightly) different methods. An example to the titration task of determining the acidity of a sample you find here.
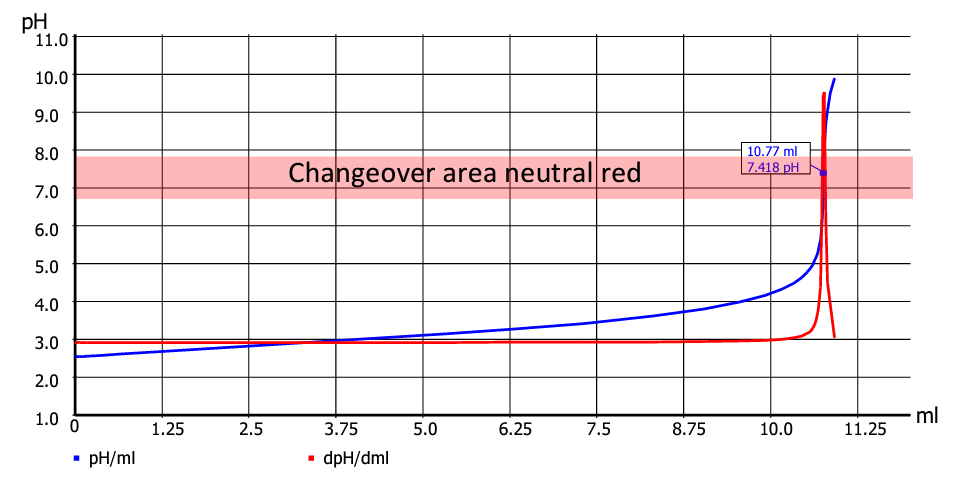
Fig. 3: Changeover area and equivalence point with a suitable indicator (Titration-Fibel Page 49).
Content Titration Handbook (an excerpt of the 192 pages of compact titration knowledge)
1. Basics of titration
- Titration reactions (e.g. Acid-base titration, Precipitation titration, complexometric titration, Redox titration, charge transfer titration, chemical, visual, Potentiometric, Biamperometric, Photometric, conductometric, thermometric
- Titration types (Direct titration, back titration, Indirect titration, substitution titration, phase transfer titration)
- Overview of the used methods
2. Volume measurement devices, manual and automatic titration
- Volume measurement devices in the laboratory
- Verification of the correct volume
- Comparison of manual and automatic titration
3. Sample handling
4. Sensors and reagents
5. Titration parameters and calculations
- Response behavior of the electrode and speed
- Definition of the titration end
- Evaluation of the titration
6. Applications
- Acid-base titrations
- Argentometric titrations
- Potentiometric redox titrations
- … and some more
7. Photometric titrations
9. Verification of the titration
Further information can be found in the titration catalog

Find here all components for a manual or automated titration with our high-precision software Titri Soft 3.5 for your use in the laboratory. Learn more about all products in the laboratory environment: coulometric and volumetric titrators, burettes, sample changers, the matching software and the corresponding titration electrodes and all accessories around titration.
Learn more
Find answers in the comprehensive titration handbook
 This is an excerpt from the titration handbook. On 192 pages it offers a compact introduction to the theory and practice of titration. If you are interested, you are welcome to download the practical guide as a PDF or request it as a brochure. And in our database you will find numerous applications for your titration for download. A concrete application example of the determination of acid in bases is described in the blogarticle "how to get correct and reproducible results in titration?". Our titration expert answers further questions from our customers in the blog article FAQ on titration.
This is an excerpt from the titration handbook. On 192 pages it offers a compact introduction to the theory and practice of titration. If you are interested, you are welcome to download the practical guide as a PDF or request it as a brochure. And in our database you will find numerous applications for your titration for download. A concrete application example of the determination of acid in bases is described in the blogarticle "how to get correct and reproducible results in titration?". Our titration expert answers further questions from our customers in the blog article FAQ on titration.
Further questions are answered by our expert in the blog article FAQ Titration. Helpful tips for your application area, you can read in our other blog articles: